


On the thermal expansion of the tetragonal phase of MAPbI3 and MAPbBr3
 0
0  ,
, Abstract
Based on previously published research, the structural response of the tetragonal hybrid perovskite crystal structure of MAPbX3 [MA: [CH3NH3]+, methylammonium; X = I, Br] to thermal expansion is reviewed here. From an averaged crystal structure perspective, the tetragonal perovskite structure of MAPbI3 and MAPbBr3, based on diffraction data, shows apparent Pb-X bond length shortening and apparent shrinkage of the [PbX6] octahedra with increasing temperature. At the same time, these apparent observations, and hence the thermal expansion, are related to the progressive phase transformation towards the cubic structure, as the lattice parameters respond to a shear stress that couples to the order parameters, and this coupling is predicted by group theory and thus aims to explain precisely the apparent negative thermal expansion-like effects. A different picture emerges for the thermal expansion when considering the very localized structure, since neither a shortening of the Pb-X bond lengths nor a shrinking of the [PbX6] octahedra is observed with pair distribution function analysis, and the presence of orthorhombic short-range order in the tetragonal and cubic perovskite structures is assumed in published studies. The compared extended X-ray absorption fine structure studies, which also map the local structure and provide the “true” bond distance, show no lead-halide bond length shortening with temperature. The perpendicular mean square relative displacement has been determined. Therefore, a comparison of the tension and bond expansion effects in both perovskites can be made. In the orthorhombic phase of MAPbI3 and MAPbBr3, positive expansion and negative tension of the lead-halide bond are almost balanced. After transitioning to the tetragonal phase, the equilibrium shifts toward negative tension. This suggests that both hybrid perovskites have tighter lead-halide bonds and less rigid [PbX6] octahedra in the tetragonal phase than in the low temperature perovskite crystal structure.

Keywords
INTRODUCTION
Due to their optoelectronic properties (high optical absorption coefficients, narrow-band bright photoluminescence, tuneable bandgaps, low exciton binding energies, and long-range carrier diffusion)[1-3], hybrid perovskites are promising materials for applications in photovoltaics[4,5]. Hybrid perovskites of the general form MAPbX3 (MA: [CH3NH3]+, methylammonium; X = I, Br) such as MAPbI3 and MAPbBr3, undergo phase transformations from the cubic high-temperature aristo-type structure (space group Pm
In a selective short review, the thermal expansion of the tetragonal phase of MAPbI3 and MAPbBr3 is discussed here on the basis of published work in which results from X-ray diffraction (XRD) and EXAFS are combined[6,7]. Based on the results of the structural investigations on the temperature-dependent XRD data of MAPbI3 and MAPbBr3, the thermal expansion in the tetragonal phases is analyzed and described by linear thermal expansion coefficients. The temperature dependence of these thermal expansion coefficients is then quantified for MAPbI3. For both components, the phase transformation from the tetragonal to the cubic perovskite structure is described by the corresponding order and strain parameters. Based on the PDF analysis of MAPbBr3 by Bernasconi et al., the thermal expansion is discussed from a local order perspective[16]. Finally, the results of temperature-dependent EXAFS analyses of MAPbI3 and MAPbBr3 are compared and discussed.
DISCUSSION
Apparent shrinkage of [PbX6] octahedra and Pb-X bond lengths changes in the tetragonal phases of MAPbI3 and MAPbBr3
Two previous temperature-dependent crystallographic investigations of MAPbI3 from Schuck et al.[6] and of MAPbBr3 from Weadock et al.[7] are compared and discussed in relation to the observed volume changes of the [PbX6] octahedra and the changes in Pb-X bond lengths. In the following, we will focus on the tetragonal phase [Figure 1] and the transition to the cubic structure by looking at the behavior of the [PbX6] octahedra network in dependence on temperature [Figure 2]. Powder samples of MAPbI3 were measured by Schuck et al. using synchrotron XRD as a function of temperature, and the resulting diffractograms were refined using the Rietveld method. The MA molecule geometry was taken from the previous density functional theory (DFT) optimization and treated as rigid[35]. The results of the structure refinements of MAPbI3[6] agree with previous temperature-dependent studies[36-38].

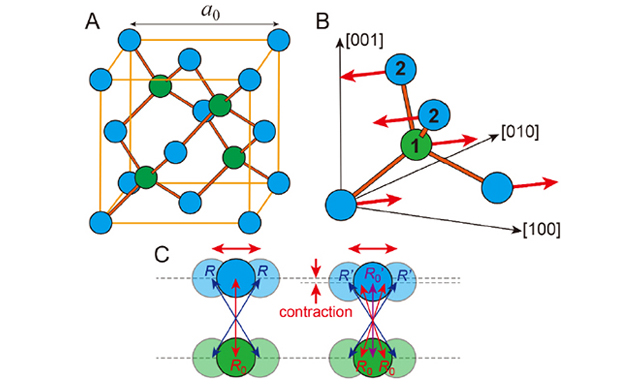
Figure 1. Detail of the tetragonal room-temperature structure of MAPbI3 with its large iodine anisotropic temperature factors (ADPs) (their flat shape indicates a transverse displacement perpendicular to the Pb-I-Pb bonds and in the direction of the MA molecule), the coordination polyhedra of the MA molecule [MAI12], a cuboctahedron (light yellow), and one [PbI6] octahedra (light gray)[37]. The anisotropic shape and orientation of the halide ADPs are partly due to the strongly anharmonic metal-halide bond (see EXAFS section) and not only to the hydrogen bond between the MA and the halide atoms (formed between the jump rotations). In this representation of a single MA molecule, neither the 4-fold rotational axis nor the 2-fold rotational axis of the I4/mcm space group symmetry seems to exist for the molecule. However, since the MA molecule jump rotates[8] and there are, on average, eight MA molecule orientations in the cage, an average structure with I4/mcm symmetry is observed in diffraction.

Figure 2. (A) The [PbI6] octahedra network of MAPbI3 for the cubic high-temperature structure at 350 K[36] (top) and the tetragonal room-temperature structure at 300 K[37] (bottom). (B) Pb-Xa-Pb bond angle (octahedra tilt) of MAPbI3 (red) and MAPbBr3 (blue) in the tetragonal (solid symbols) and cubic phase (open symbols). Besides the data from Schuck et al.[6] and Weadock et al.[7], values for the cubic phase at 350 K of MAPbI3 from Whitfield et al. are also given[36]. Adapted with permission from Schuck et al.[6]. Copyright 2022 American Chemical Society.
The volume of MAPbI3 increases with temperature over the whole temperature range (volumetric thermal expansion coefficient αV, orthorhombic = 90 × 10-6 K-1 and αV, tetragonal = 119 × 10-6 K-1). Based on the lattice constants for a pseudo-cubic cell from Schuck et al., a negative linear thermal expansion coefficient of

Figure 3. Temperature-dependent lattice constants for the tetragonal (pseudo-cubic) and cubic unit cell for (A) MAPbI3 (cubic at

Figure 4. (A) Averaged Pb-X bond lengths expansion and (B) volumetric expansion of MAPbI3 (red) and MAPbBr3 (blue) in the tetragonal (solid symbols) and cubic phase (open and half-open symbols), the phase transformation temperature is indicated in each case (tetragonal to cubic). Besides the data from Schuck et al.[6] and Weadock et al.[7], values for the cubic phase of MAPbI3 are also given from Whitfield et al.[36] (left half-open symbols) and Stoumpos et al.[38] (right half-open symbols). (B) In addition to the volume changes for the [PbX6] octahedra (squares), the volume changes for the total volume (diamond symbols) and the volume changes for the [MAX12] cuboctahedra (circles) are also given. Note: Both Pb-X bond lengths, i.e., the one along tetragonal c (Pb-Xb in Figure 2A) and the one in the tetragonal a-b plane (Pb-Xa in Figure 2A), become smaller in the tetragonal phase with increasing temperature. Both bond lengths (Pb-Xa and Pb-Xb) for MAPbI3 and MAPbBr3 are within the error bars of the averaged value Pb-X. Adapted with permission from Schuck et al.[6]. Copyright 2022 American Chemical Society.
Temperature-dependent linear thermal expansion coefficients and the Grüneisen tensor
The linear thermal expansion coefficients calculated for MAPbI3 from the data of Schuck et al. differ significantly from the values of Jacobsson et al.[6,27]. This is not surprising since one linear fit obviously cannot describe the entire temperature range of the tetragonal phase. For this reason, the temperature dependence of the linear thermal expansion coefficients is investigated using the tetragonal lattice constants of Whitfield et al. [the lattice constants from temperature-dependent synchrotron powder XRD measurements of Whitfield et al. (figure 6C in Ref.[36]) have been digitized by us and are shown here in Figure 5A][36]. The temperature dependence of the linear thermal expansion coefficients [Figure 5B] can be approximately described for the PTE α[100] with a linear behavior; an increase towards the cubic phase up to 97 × 10-6 K-1 is obtained. The NTE α[001] can be described with a third-order polynomial and reaches a maximum value of -88 × 10-6 K-1 towards the cubic phase. Due to the temperature dependence of the two linear thermal expansion coefficients α[001] and α[100], the linear thermal expansion coefficient α[averaged] determined from the averaged tetragonal lattice constant aaveraged = ((2*atet)+ctet)/3 is more suitable for describing the thermal expansion as it has almost no temperature dependence. The value given by

Figure 5. (A) Temperature-dependent tetragonal (pseudo-cubic) lattice constants according to Whitfield et al.[36] (black) and from Schuck et al.[6] (red). (B) Temperature dependence of the linear thermal expansion coefficients α[100], α[001], and α[averaged] based on the lattice constants of Whitfield et al. [displayed in (A)][36]. α[100]: linear behavior; α[001]: third-order polynomial (solid lines). Adapted with permission from Schuck et al.[6]. Copyright 2022 American Chemical Society.

Figure 6. (A) Temperature-dependent values of
In the case of the tetragonal crystal structure of hybrid perovskites, two independent Grüneisen parameters, γ[100] and γ[001], are related to the principal linear thermal expansion coefficients α[100] and α[001] via (according to Schorr et al.[43]):
where Vm is the molar volume, Cp is the molar specific heat at constant pressure, and the cij are the adiabatic elastic stiffness coefficients in this Grüneisen tensor (according to Haussühl and Kitaigorodskii)[44]. Using the tabulated cij values[45], a negative Grüneisen parameter γ[001] can only be expected near the phase transition to the cubic phase above 307 K [Figure 6A]. At lower temperatures, γ[001] is positive and the Grüneisen parameter γ[100] is always positive for MAPbI3 [Figure 6B].
Tetragonal to the cubic phase transition
According to Whitfield et al., the phase transition from the tetragonal to the cubic crystal structure in MAPbI3 and MAPbBr3 can be described by the temperature-dependent change of three degrees of freedom in the tetragonal structure: the rotational distortion caused by the change of the Pb-Ia-Pb angle (R4+), the strain caused by the thermal expansion (GM1+), and the strain caused by the tetragonal distortion (GM3+)[36].
The temperature-dependent changes of the three degrees of freedom were analyzed for MAPbI3 and MAPbBr3 using ISODISTORT (an internet-server tool for exploring structural phase transitions)[46] and are shown in Figure 7. The thermal expansion in the tetragonal phase discussed in the previous two sections is related to the progressive phase transformation towards the cubic structure, since the lattice parameters respond to a shear stress coupled to the order parameters (analyzed with ISODISTORT), and this coupling is predicted by a standard Landau theory treatment. The amplitude of the R4+ rotational mode, which is directly proportional to the cubic-tetragonal order parameter, can be fitted by the power law

Figure 7. ISODISTORT analysis[46]. The phase transition from tetragonal to cubic, based on the data from Schuck et al.[6] and Liu et al.[47] for MAPbI3 and based on the data from Weadock et al.[7] for MAPbBr3 can be described by the temperature-dependent changes of the three degrees of freedom in the tetragonal structure: rotational distortion mode R4+ (blue color, legend on the right) and the two strain mode amplitudes (black color, legend on the left) GM1+ (thermal expansion) and GM3+ (tetragonal distortion (c-a)/c). Fit of the order parameter R4+ for MAPbI3 based on the data from Schuck et al.[6] (blue dashed line) and fit for the data from Liu et al.[47] (blue dotted line), additional also the R4+ fit from Whitfield et al.[36] (figure 8A in Ref.[36]) is reproduced (Tc = 329.95 K and β = 0.26, solid blue line). Fit of the order parameter R4+ for MAPbBr3 based on the data from Weadock et al. (blue dash-dotted line)[7].
Local structure of MAPbI3 and MAPbBr3 with atomic pair distribution function (PDF) analysis
The local atomic structure can be investigated by analyzing the total scattering from diffraction experiments[48,49], which, in contrast to XRD or neutron powder diffraction methods, use much shorter
In the temperature-dependent pair correlation functions G(r) of MAPbI3, it is, first of all, noticeable, as already noted by Whitfield et al., that in a range up to about 4 Å, no discontinuous changes occur in the range from 10 to 350 K, but only a gradual broadening of G(r) towards high temperatures (see figure 4 in Ref.[36]), which is also especially the case in the range of the phase transformation from the orthorhombic to the tetragonal structure (o/t)[36]. This range up to 4 Å, which does not change discontinuously, corresponds to the Pb-X bond distances. The situation is different for the range from 4 Å to larger distances, where a discontinuous change in G(r) in the temperature range of the o/t phase transformation is observed.
A very similar behavior can also be observed in the data available for MAPbBr3. Here, there is also no discontinuous change at small distances in the region of the o/t phase transformation (see figure 1 in

Figure 8. Short-range fit (r range = 1-10 Å) by using (A) the tetragonal MAPbBr3 perovskite structure (I4/mcm) and (B) the orthorhombic MAPbBr3 perovskite structure (Pnma). For the PDF fit (with PDFgui 1.0), the data from Bird et al.[17] at 200 K was used together with the fixed refinement results at 200 K from Bernasconi et al.[16]. In the PDF fit, only the scale and Uiso were refined.

Figure 9. Results of the PDF analysis by Bernasconi et al.[16] using the G(r) range 1-10 Å with orthorhombic Pnma symmetry. (A) orthorhombic perovskite structure at 200 K; (B) orthorhombic lattice constants (pseudo-cubic); (C) averaged Pb-Br bond lengths expansion and (D) volumetric expansion of MAPbBr3. Adapted with permission from Bernasconi et al.[16]. Copyright 2017 American Chemical Society.
Analysis of the anharmonic properties of the lead-halide bond with EXAFS
EXAFS spectroscopy is based on the ionization of an atom when an X-ray quantum is absorbed. This releases an electron whose kinetic energy depends on the energy of the X-rays. The released electron propagates as a matter wave and is scattered by neighboring atoms. Depending on the wavelength of the electron, constructive or destructive interference occurs between the outgoing wave and the backscattered waves. Because the energy of the X-rays is varied, the energy of the released electrons also changes and thus the corresponding wavelength of the electrons. This leads to alternating constructive and destructive interference and, thus, to a change in X-ray absorption depending on the energy, which corresponds to the fine structure of the X-ray absorption spectrum. These X-ray absorption changes can be measured in the energy range from just above the X-ray absorption edge up to a few hundred electron volts above it, i.e., in the upper range of the X-ray absorption edge (hence the term “extended” in EXAFS). From the shape and strength of the X-ray absorption changes, it is possible to deduce at what distance from the ionized atom it is scattered and to what extent, i.e., a so-called radial distribution function is obtained via Fourier transformation of the X-ray absorption “fine structure” data after normalization and background subtraction (for example with the Athena software)[52]. By comparing the scattering of electrons on and between neighboring atoms with simulations (for example, with the Artemis software)[6,52,53], the atomic distances of atom pairs REXAFS can be determined with high accuracy. However, the radial distribution function not only contains information about the atomic distances but also about the distance variations of the respective atom pairs, which corresponds to the parallel MSRD (EXAFS Debye-Waller factor or C2). Since the lifetime of the EXAFS photoelectron (~10-15 s) is very short compared to the atomic vibrations (~10-12 s), and, at the same time, the limited mean free path of the EXAFS photoelectron is restricted to the local environment of the investigated atom, the anharmonic properties of the investigated bond pair can be studied very well[34]. The relative ΔC2 (T) was fitted with an Einstein behavior by Schuck et al.[6] to determine parallel MSRD (T)[54] of MAPbI3:
(µ: reduced mass, ΘE: Einstein temperature). The resulting parallel MSRD (T) is shown in Figure 10A. This figure also shows the parallel MSRD (T) for MAPbBr3, which was interpreted by Weadock et al. using both a correlated Debye model and an Einstein behavior [in the Debye model, a distinction was made between a low-temperature (ΘD,LT = 148 K) and a high-temperature range (ΘD,LT = 167 K)][7]. Weadock et al. noted that the Einstein temperatures for MAPbI3 and MAPbBr3 increase with decreasing reduced mass

Figure 10. Temperature-dependent MSRDs: (A) parallel, (B) perpendicular and (C) γEXAFS. Red: MAPbI3 (Schuck et al.[6]); blue: MAPbBr3 (Weadock et al.[7]). Solid lines: Einstein fits. In (B) the parallel MSRD values for MAPbI3 were fitted with two Einstein temperatures, one for the orthorhombic low-temperature range up to 160 K, the other for the tetragonal high-temperature range up to 260 K (here with an additional static component of 0.05). This procedure also leads to a better explanation of the experimental values for the description of the γEXAFS dependence in (C). Adapted with permission from Schuck et al.[6]. Copyright 2022 American Chemical Society.
[1 N/m = 1/16.022 eV/Å2], [1 THz = 47.9924 K], and νE: Einstein frequency [THz] [Table 1].
Einstein temperatures and frequencies for parallel (ΘE||, νE||) and perpendicular MSRD (
| MAPbI3[6] | MAPbBr3[7] | |||
| µ | 78.702 | 57.666 | ||
| Pb-X bond-stretching | ||||
| ΘE|| [K] | 97.0 (3) | 117.3 (4) | ΘD|| [K] | LT: 148 (4); HT: 167 (5) |
| νE|| [THz] | 2.021 (5) | 2.444 (6) | νD|| [THz] | LT: 3.08 (8); HT: 3.48 (10) |
| k|| [eV/Å2] | 1.315 (3) | 1.410 (6) | k|| [eV/Å2] | LT: 1.43 (3); HT: 1.87 (5) |
| Pb-X bond-bending | ||||
| LT: 46.9 (8); HT: 50 (1) | HT: 46 (1) | |||
| LT: 0.98 (2); HT: 1.04 (2) | HT: 0.96 (2) | |||
| k⊥ [eV/Å2] | LT: 0.309 (6); HT: 0.348 (7) | HT: 0.218 (5) | ||
From the temperature-dependent relative interatomic distances ΔREXAFS and the averaged relative interatomic distances ΔRXRD resulting from the synchrotron XRD [Figure 11], Schuck et al. determine the relative “perpendicular MSRD” (

Figure 11. Relative Pb-X expansion of MAPbI3 from Schuck et al.[6] (red) and MAPbBr3 (Weadock et al.[7]) (blue). Positive bond expansion ΔREXAFS (solid upward triangles) and negative tension effect (solid downward triangles) result in the overall crystallographic expansion ΔRXRD (open circles). Values for the cubic perovskite phase of MAPbI3 are also given from Whitfield et al.[36] (open red star symbol). Adapted with permission from Schuck et al.[6]. Copyright 2022 American Chemical Society.
The following relationship applied:[55]
The absolute values of the perpendicular MSRD for MAPbI3 were then obtained, analogously to the parallel MSRD, through fitting of
Analogous to k0, the corresponding perpendicular effective force constant k⊥ (bond-bending effective force constant) from the correlated Einstein model could be calculated for MAPbI3 [Table 1][6]. Analogous to the procedure for MAPbI3, the relative perpendicular MSRD and the bond-bending effective force constant were also determined for MAPbBr3 from the available data[7]. For MAPbBr3, this was possible only for the tetragonal and cubic phases since the values determined for ΔREXAFS and ΔRXRD in the orthorhombic phase [Figure 11], based on the results of Weadock et al., lead only to unphysical values for the relative perpendicular MSRD (here, the digitization of the data from Weadock et al. could not be carried out with sufficient accuracy for the orthorhombic phase)[7]. However, since the phase transformation from tetragonal to cubic is being discussed here, this is not limiting. The ratio γEXAFS = perpendicular MSRD/parallel MSRD describes the degree of anisotropy of the relative vibrations [Figure 10C][34]. The uncertainties pointed out by Schuck et al. may be of a similar (if not larger) magnitude in the behavior of MAPbBr3 shown here, so that the anisotropy of the relative vibrations and the perpendicular MSRD of MAPbI3 and MAPbBr3 are unlikely to differ significantly. However, perpendicular MSRD
Also, an attempt is made here to establish correlations between the relative atomic vibrations obtained from EXAFS studies on one hand and the behavior of optical phonons on the other. In addition, the Grüneisen parameters of individual phonon modes (also known as phonon deformation potentials) are of interest as a probe of the anharmonicity associated with phonon-phonon scattering and heat transport[59,62,63]. Two recent publications investigated the temperature dependence of optical phonons using infrared (IR) and terahertz measurements of MAPbI3 and MAPbBr3[64,65]. Two optical phonons at around 0.9 and 1.8 THz have been reported from Boldyrev et al. in MAPbI3 single crystals using temperature-dependent (figure 5 in
CONCLUSION
The review of the results from the analysis of the averaged crystal structure with XRD diffraction methods shows temperature-dependent changes in the tetragonal phase with an apparent shortening of the Pb-X bonds and an apparent shrinkage of the [PbX6] octahedra in both MAPbI3 and MAPbBr3 with increasing temperature up to the cubic phase. The thermal expansion can best be described with the linear thermal expansion coefficient α[averaged] for the averaged tetragonal lattice constant aaveraged = ((2*atet)+ctet)/3 since the lowest temperature dependence occurs for this expansion coefficient. Using the Grüneisen tensor, positive Grüneisen parameters γ[001] and γ[100] can be expected for nearly the complete temperature range of the tetragonal phase of MAPbI3 (with the exception of γ[001] in a small temperature range close to the cubic phase transition above 307 K). The analysis of the tetragonal/cubic phase transformation using the standard Landau theory treatment seems to show a certain variance in the determination of the order parameters, but this can probably largely be explained by the quality of the underlying structure refinements. From the point of view of the local structure, it is remarkable that the thermal expansion of the very local arrangement of the lead halide octahedra is apparently unaffected by the tetragonal/cubic phase transformation. The temperature-dependent changes in the range of 150-300 K of the Pb-Br bond distances determined for MAPbBr3 by Bernasconi et al. from PDF refinements in the range of 1-10 Å using the orthorhombic Pnma perovskite crystal structure are of approximately the same order of magnitude as the bond expansion values ΔREXAFS determined by Weadock et al. with EXAFS[7,16]. Tension effects are observed with EXAFS in the tetragonal phase for both MAPbI3 and MAPbBr3. The positive bond extension and the negative tension effects of the lead-halide bond are approximately the same in MAPbI3 (and presumably also in MAPbBr3) in the orthorhombic phase. After the transition to the tetragonal phase, the equilibrium shifted in favor of the negative tension effects in both MAPbI3 and MAPbBr3. Further analysis of similarities between EXAFS relative atomic vibrations and optical phonon studies could be a rewarding way to better understand the thermal expansion in the tetragonal perovskite structure of MAPbI3 and MAPbBr3. This brief overview of the thermal expansion of MAPbI3 and MAPbBr3 can hopefully contribute to a more comprehensive description that includes all relevant inorganic halide perovskites and the discussion of NTE coefficients for ferroelectric oxide perovskites.
DECLARATIONS
Authors’ contributions
Contributed equally to this article: Schuck G, Többens DM, Schorr S
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author (s) 2024.
REFERENCES
1. Chouhan L, Ghimire S, Subrahmanyam C, Miyasaka T, Biju V. Synthesis, optoelectronic properties and applications of halide perovskites. Chem Soc Rev 2020;49:2869-85.
2. Pérez-Fidalgo L, Xu K, Charles BL, et al. Anomalous electron-phonon coupling in cesium-substituted methylammonium lead iodide perovskites. J Phys Chem C 2023;127:22817-26.
3. Spera EL, Pereyra CJ, Gau DL, Berruet M, Marotti RE. Excitonic optical properties of CH3NH3PbI3 perovskite and its dependence with temperature. MRS Adv 2024;9:39-44.
4. Jošt M, Kegelmann L, Korte L, Albrecht S. Monolithic perovskite tandem solar cells: a review of the present status and advanced characterization methods toward 30% efficiency. Adv Energy Mater 2020;10:1904102.
5. Duan L, Walter D, Chang N, et al. Stability challenges for the commercialization of perovskite-silicon tandem solar cells. Nat Rev Mater 2023;8:261-81.
6. Schuck G, Többens DM, Wallacher D, Grimm N, Tien TS, Schorr S. Temperature-dependent EXAFS measurements of the Pb L3-edge allow quantification of the anharmonicity of the lead-halide bond of chlorine-substituted methylammonium (MA) lead triiodide. J Phys Chem C 2022;126:5388-402.
7. Weadock NJ, Mackeen C, Qin X, et al. Thermal contributions to the local and long-range structural disorder in CH3NH3PbBr3. PRX Energy 2023;2:033004.
8. Schuck G, Lehmann F, Ollivier J, Mutka H, Schorr S. Influence of chloride substitution on the rotational dynamics of methylammonium in MAPbI3-xClx perovskites. J Phys Chem C 2019;123:11436-46.
9. Miyata K, Atallah TL, Zhu XY. Lead halide perovskites: crystal-liquid duality, phonon glass electron crystals, and large polaron formation. Sci Adv 2017;3:e1701469.
10. Tailor NK, Satapathi S. Crystalline-liquid duality of specific heat in halide perovskite semiconductor. Scr Mater 2023;223:115061.
11. Adams DJ, Churakov SV. Classification of perovskite structural types with dynamical octahedral tilting. IUCrJ 2023;10:309-20.
12. Liang X, Klarbring J, Baldwin WJ, Li Z, Csányi G, Walsh A. Structural dynamics descriptors for metal halide perovskites. J Phys Chem C Nanomater Interfaces 2023;127:19141-51.
13. Weadock NJ, Sterling TC, Vigil JA, et al. The nature of dynamic local order in CH3NH3PbI3 and CH3NH3PbBr3. Joule 2023;7:1051-66.
14. Beecher AN, Semonin OE, Skelton JM, et al. Direct observation of dynamic symmetry breaking above room temperature in methylammonium lead iodide perovskite. ACS Energy Lett 2016;1:880-7.
15. Page K, Siewenie JE, Quadrelli P, Malavasi L. Short-range order of methylammonium and persistence of distortion at the local scale in MAPbBr3 hybrid perovskite. Angew Chem Int Ed 2016;55:14320-4.
16. Bernasconi A, Malavasi L. Direct evidence of permanent octahedra distortion in MAPbBr3 hybrid perovskite. ACS Energy Lett 2017;2:863-8.
17. Bird TA, Chen J, Songvilay M, et al. Large dynamic scissoring mode displacements coupled to band gap opening in hybrid perovskites. arXiv 2021. Available from: https://arxiv.org/abs/2108.05751 [Last accessed on 7 Aug 2024].
18. Simenas M, Gagor A, Banys J, Maczka M. Phase transitions and dynamics in mixed three- and low-dimensional lead halide perovskites. Chem Rev 2024;124:2281-326.
19. Kutes Y, Ye L, Zhou Y, Pang S, Huey BD, Padture NP. Direct observation of ferroelectric domains in solution-processed CH3NH3PbI3 perovskite thin films. J Phys Chem Lett 2014;5:3335-9.
20. Bari M, Bokov AA, Ye Z. Ferroelastic domains and phase transitions in organic-inorganic hybrid perovskite CH3NH3PbBr3. J Mater Chem C 2021;9:3096-107.
21. Bari M, Bokov AA, Leach GW, Ye Z. Ferroelastic domains and effects of spontaneous strain in lead halide perovskite CsPbBr3. Chem Mater 2023;35:6659-70.
22. Wilson JN, Frost JM, Wallace SK, Walsh A. Dielectric and ferroic properties of metal halide perovskites. APL Mater 2019;7:010901.
23. Breternitz J. The “ferros” of MAPbI3: ferroelectricity, ferroelasticity and its crystallographic foundations in hybrid halide perovskites. Cryst Mater 2022;237:135-40.
24. Ambrosio F, De Angelis F, Goñi AR. The ferroelectric-ferroelastic debate about metal halide perovskites. J Phys Chem Lett 2022;13:7731-40.
25. Zheng W, Wang X, Zhang X, et al. Emerging halide perovskite ferroelectrics. Adv Mater 2023;35:e2205410.
26. Haeger T, Heiderhoff R, Riedl T. Thermal properties of metal-halide perovskites. J Mater Chem C 2020;8:14289-311.
27. Jacobsson TJ, Schwan LJ, Ottosson M, Hagfeldt A, Edvinsson T. Determination of thermal expansion coefficients and locating the temperature-induced phase transition in methylammonium lead perovskites using X-ray diffraction. Inorg Chem 2015;54:10678-85.
28. Bozec Y, Kaang S, Hine P, Ward I. The thermal-expansion behaviour of hot-compacted polypropylene and polyethylene composites. Composit Sci Technol 2000;60:333-44.
29. Becker P, Scyfried P, Siegert H. The lattice parameter of highly pure silicon single crystals. Z Physik B Condens Matter 1982;48:17-21.
30. Ge C, Hu M, Wu P, et al. Ultralow thermal conductivity and ultrahigh thermal expansion of single-crystal organic-inorganic hybrid perovskite CH3NH3PbX3 (X = Cl, Br, I). J Phys Chem C 2018;122:15973-8.
31. Zhou Y, Guo Z, Qaid SMH, Xu Z, Zhou Y, Zang Z. Strain engineering toward high-performance formamidinium-based perovskite solar cells. Solar RRL 2023;7:2300438.
33. Fornasini P, Grisenti R. On EXAFS debye-waller factor and recent advances. J Synchrotron Rad 2015;22:1242-57.
34. Sanson A. EXAFS spectroscopy: a powerful tool for the study of local vibrational dynamics. Microstructures 2021;1:2021004.
35. Schuck G, Többens DM, Koch-müller M, Efthimiopoulos I, Schorr S. Infrared spectroscopic study of vibrational modes across the orthorhombic-tetragonal phase transition in methylammonium lead halide single crystals. J Phys Chem C 2018;122:5227-37.
36. Whitfield PS, Herron N, Guise WE, et al. Structures, phase transitions and tricritical behavior of the hybrid perovskite methyl ammonium lead iodide. Sci Rep 2016;6:35685.
37. Franz A, Többens DM, Schorr S. Interaction between cation orientation, octahedra tilting and hydrogen bonding in methylammonium lead triiodide. Cryst Res Technol 2016;51:534-40.
38. Stoumpos CC, Malliakas CD, Kanatzidis MG. Semiconducting tin and lead iodide perovskites with organic cations: phase transitions, high mobilities, and near-infrared photoluminescent properties. Inorg Chem 2013;52:9019-38.
39. Celeste A, Capitani F. Hybrid perovskites under pressure: present and future directions. J Appl Phys 2022;132:220903.
40. Szafrański M, Katrusiak A. Mechanism of pressure-induced phase transitions, amorphization, and absorption-edge shift in photovoltaic methylammonium lead iodide. J Phys Chem Lett 2016;7:3458-66.
41. Gil-González E, Pérez-Maqueda LA, Sánchez-Jiménez PE, Perejón A. Paving the way to establish protocols: modeling and predicting mechanochemical reactions. J Phys Chem Lett 2021;12:5540-6.
42. Whitfield PS, Herron N, Guise WE, et al. Correction: Corrigendum: structures, phase transitions and tricritical behavior of the hybrid perovskite methyl ammonium lead iodide. Sci Rep 2017;7:42831.
43. Schorr S, Sheptyakov D. Low-temperature thermal expansion in sphalerite-type and chalcopyrite-type multinary semiconductors. J Phys Condens Matter 2008;20:104245.
44. Haussühl S. Kristallphysik; Weinheim, Germany: Physic-Verlag; 1983.
45. Feng J. Mechanical properties of hybrid organic-inorganic CH3NH3BX3 (B = Sn, Pb; X = Br, I) perovskites for solar cell absorbers. APL Mater 2014;2:081801.
46. Campbell BJ, Stokes HT, Tanner DE, Hatch DM. ISODISPLACE: a web-based tool for exploring structural distortions. J Appl Cryst 2006;39:607-14.
47. Liu J, Du J, Phillips AE, Wyatt PB, Keen DA, Dove MT. Neutron powder diffraction study of the phase transitions in deuterated methylammonium lead iodide. J Phys Condens Matter 2022;34:145401.
48. Egami T, Billinge SJL. Underneath the bragg peaks: structural analysis of complex materials; Oxford: Elsevier; 2003.
49. Billinge SJ. Nanoscale structural order from the atomic pair distribution function (PDF): there’s plenty of room in the middle. J Solid State Chem 2008;181:1695-700.
50. Bird TA, Herlihy A, Senn MS. Symmetry-adapted pair distribution function analysis (SAPA): a novel approach to evaluating lattice dynamics and local distortions from total scattering data. J Appl Cryst 2021;54:1514-20.
51. Bird TA, Woodland-Scott J, Hu L, et al. Anharmonicity and scissoring modes in the negative thermal expansion materials ScF3 and CaZrF6. Phys Rev B 2020;101:064306.
52. Ravel B, Newville M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Rad 2005;12:537-41.
53. Bunker G. Application of the ratio method of EXAFS analysis to disordered systems. Nucl Instrum Methods Phys Res 1983;207:437-44.
54. Bunker G. Introduction to EXAFS; Cambridge: Cambridge University Press; 2010.
55. Fornasini P, a Beccara S, Dalba G, et al. Extended X-ray-absorption fine-structure measurements of copper: local dynamics, anharmonicity, and thermal expansion. Phys Rev B 2004;70:174301.
56. Fornasini P, Grisenti R. The coefficient of bond thermal expansion measured by extended X-ray absorption fine structure. J Chem Phys 2014;141:164503.
57. Fornasini P. Vibrational anisotropy. In: Schnohr CS, Ridgway MC, editors. X-ray absorption spectroscopy of semiconductors. Berlin: Springer; 2015. pp. 127-42.
59. Dove MT, Fang H. Negative thermal expansion and associated anomalous physical properties: review of the lattice dynamics theoretical foundation. Rep Prog Phys 2016;79:066503.
60. Dalba G, Diop D, Fornasini P, Rocca F. An EXAFS study of thermal disorder in GaAs. J Phys Condens Matter 1994;6:3599-608.
61. Dalba G, Fornasini P, Kuzmin A, Purans J, Rocca F. X-ray absorption spectroscopy study of ReO3 lattice dynamics. J Phys Condens Matter 1995;7:1199-213.
62. Talit K, Strubbe DA. Stress effects on vibrational spectra of a cubic hybrid perovskite: a probe of local strain. J Phys Chem C 2020;124:27287-99.
63. Gava V, Martinotto AL, Perottoni CA. First-principles mode Gruneisen parameters and negative thermal expansion in α-ZrW2O8. Phys Rev Lett 2012;109:195503.
64. Boldyrev KN, Anikeeva VE, Semenova OI, Popova MN. Infrared spectra of the CH3NH3PbI3 hybrid perovskite: signatures of phase transitions and of organic cation dynamics. J Phys Chem C 2020;124:23307-16.
Cite This Article
 , ... Susan Schorr
, ... Susan SchorrHow to Cite
Schuck, G.; Többens D. M.; Schorr, S. On the thermal expansion of the tetragonal phase of MAPbI3 and MAPbBr3. Microstructures. 2024, 4, 2024047. http://dx.doi.org/10.20517/microstructures.2024.33
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Related

Data & Comments
Data
0



 Like This Article 1
likes
Like This Article 1
likes













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.